(Las animaciones de esta página proceden de Tokyo
Medical University, © Hironao Numabe, M.D.)![]()
| Citogenética
Clínica
3ª Parte |
Traducido de:
Clinical Cytogenetics--Part 1 Mary Beth Dinulos, M.D. HuBio 554 Medical Genetics, Fall 1997 Dr. Marshall Horwitz, Univ. Washington Reproducido con permiso del autor |
(Las animaciones de esta página proceden de Tokyo
Medical University, © Hironao Numabe, M.D.)![]()
Nota: los términos médicos pueden no estar correctamente traducidos
G. Anomalías cromosómicas estructurales1. Equilibradas- la reorganización no produce pérdida ni ganancia de material cromosómico
- normalmente no hay efecto fenotípico (aunque es posible que el punto de ruptura interrumpa un gen)
- riesgo de gametos desequilibradosP.ej.: Inversiones
Translocaciones (recíproca y de Robertson)2. Desequilibradas
- la reorganización provoca una pérdida o una ganancia de material cromosómico
- normalmente hay efectos fenotípicos severosP.ej.: Deleciones
Duplicaciones3. Translocaciones
- intercambio de material genético entre cromosomas no homólogos
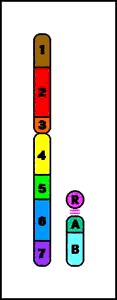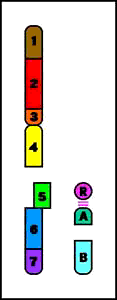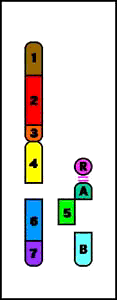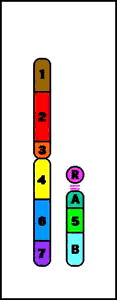
- incidencia: 1/500 individuosTranslocaciones recíprocas
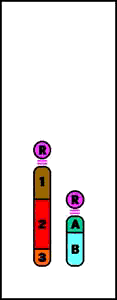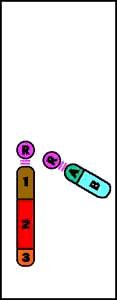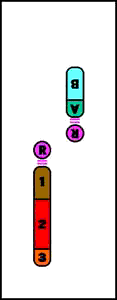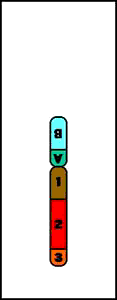
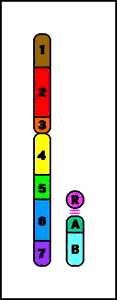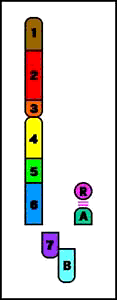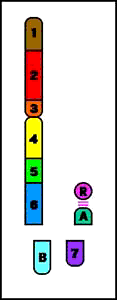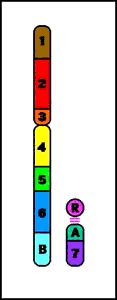 Animación que muestra las consecuencias meióticas de una translocación equilibrada- ruptura de cromosomas no homólogos con intercambio recíproco de material cromosómico (los cromosomas resultantes se denominan cromosomas derivados)
Animación que muestra las consecuencias meióticas de una translocación equilibrada- ruptura de cromosomas no homólogos con intercambio recíproco de material cromosómico (los cromosomas resultantes se denominan cromosomas derivados)- el portador de una translocación recíproca es por lo común fenotípicamente normal
- riesgo de gametos desequilibrados y fenotipo anormal en la descendencia
Translocaciones de Robertson
- los brazos largos de dos cromosomas acrocéntricos se funden por los centrómeros, con pérdida de ambos brazos cortos- restringida a los cromosomas 13,14,15, 21 y 22
- habitualmente el fenotipo es normal porque los brazos cortos de los cromosomas acrocéntricos contienen copias redundantes de los genes de RNA ribosómico (material genético no esencial)
- riesgo de fenotipo anormal en la descendencia
4. Inversiones
- resultado de dos rupturas en un cromosoma, seguidas de reinserción del fragmento en su sitio original pero en orden inverso
- incidencia: 1/1.000 individuos
- el portador de la inversión es por lo común fenotípicamente normal
- descendencia con riesgo de deleciones o duplicaciones cromosómicas y fenotipo anormal
Pericéntrica = la inversión incluye el centrómero
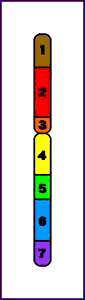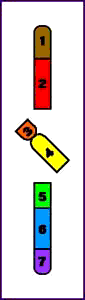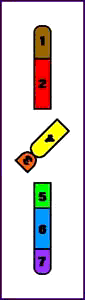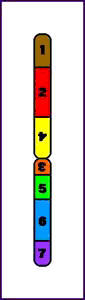
Animación que muestra las consecuencias meióticas de una inversión perincentromérica
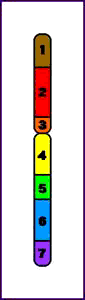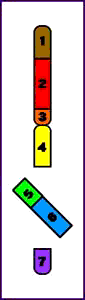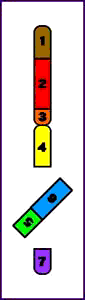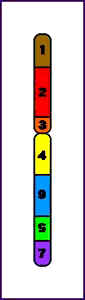
Paracéntrica = la inversión no implica al centrómero
Inversión Paracentromérica (p)
5. Deleciones
- causadas por la ruptura en un cromosoma con pérdida subsiguiente de material genético
- conduce a un fenotipo anormal (se pierden uno o varios genes)
Terminal = ruptura única que conduce a la pérdida de un extremo del cromosoma
Deleción Terminal (p)
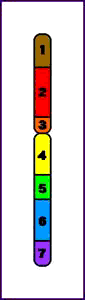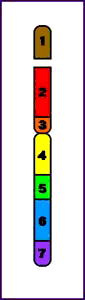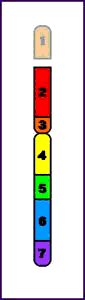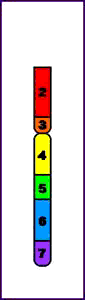
Intersticial = tienen lugar dos cortes que conducen a la pérdida del material genético entre ambos
Deleción Intersticial (q)
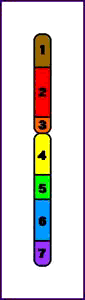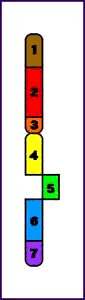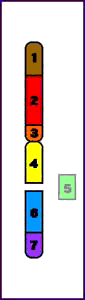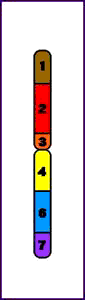
Síndrome del maullido (Cri-du-chat) (5p-)
El curioso y felino nombre de este síndrome congénito, cri du chat en su descripción original, fue idea de uno de los fundadores de la moderna citogenética: el que fuera jefe de la Unidad de Citogenética del Hospital Pediátrico Necker de París, Jérôme Lejeune (1927-1994). En una breve comunicación leída ante la Academia de Ciencias de París en 1963, el grupo
francés encabezado por Lejeune presentó los tres primeros casos conocidos de lactantes con una anomalía cromosómica por supresión o eliminación parcial del brazo corto del cromosoma 5, cuyo llanto recordaba a quien lo oía el inconfundible maullido de un gato.N.T. (F.A. Navarro, en Panace@, Vol. V, n.º 15)- incidencia: 1/50.000 nacimientos vivos
- características clínicas:
1. llanto infantil que recuerda a un gato- etiología:
2. crecimiento deficiente
3. microcefalia
4. apariencia facial peculiar
5. retraso mental profundo1. de novo (85-90%)
2. descendiente de un portador de translocación (10-15%)
Síndrome de Wolf-Hirschorn (4p-)- incidencia: 1/50.000 nacimientos vivos
- características clínicas:
1. microcefalia- etiología:
2. asimetría craneal
3. apariencia facial peculiar
4. enfermedad cardiaca congénita
5. retraso mental1. de novo (85-90%)
2. descendiente de un portador de translocación (10-15%)
IX. Condiciones para obtener un cariotipo6. Síndromes de MicrodeleciónSíndrome Velocardiofacial (Síndrome CATCH22/DiGeorge) (del22q11.2)
- incidencia: 1/10.000-1/20.000 nacimientos vivos
- características clínicas:
1. velo: paladar leporino submucoso/abierto (submucous/overt cleft palate)- etiología: deleción 22q11.2 en el 75-90% de los casos
2. cardio: defecto cardiaco conotroncal
3. facial: apariencia facial peculiar
4. problemas de aprendizaje / enfermedad psiquiátrica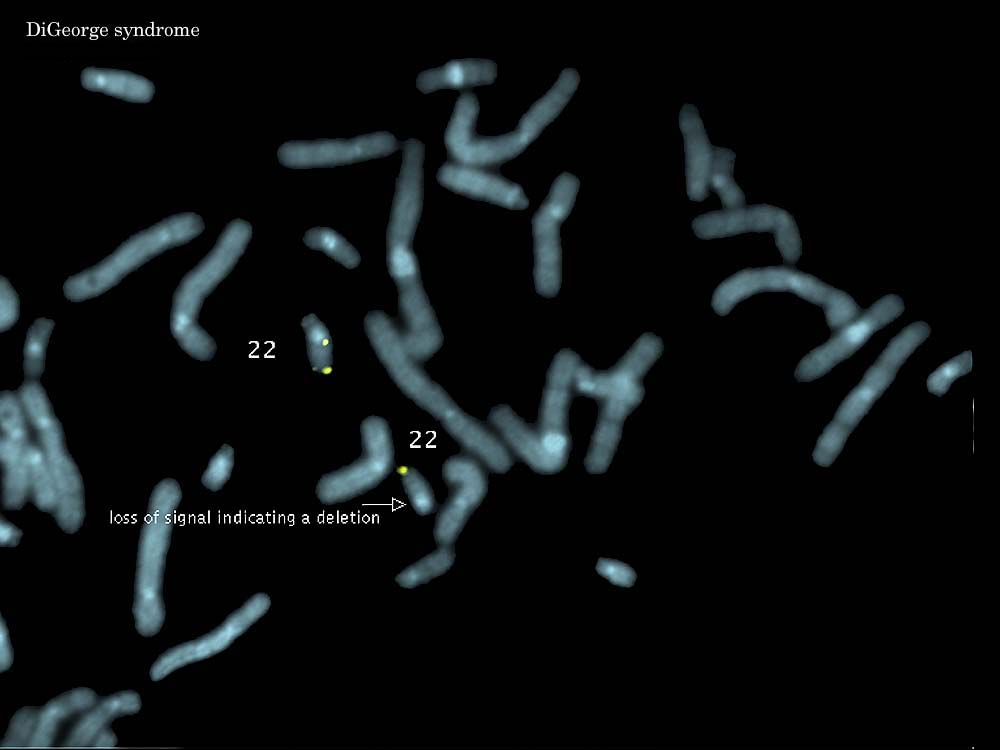 Síndrome de Prader-Willi (del15q11-q13)
Síndrome de Prader-Willi (del15q11-q13)- incidencia: 1/15.000 nacimientos vivos
- características clínicas:
1. hipotonía, alimentación pobre y falta de desarrollo en la infancia- etiología:
2. obesidad en la infancia temprana
3. hiperfagia
4. características faciales singulares
5. hipogonadismo
6. retraso mental / problemas de comportamiento1. microdeleción de 15q11-q13 paterno (~75% de los casos)- ambos cromosomas 15 son de origen materno
2. disomía uniparental materna (20-25% de los casos)
7. Duplicaciones
Duplicación (del brazo largo)
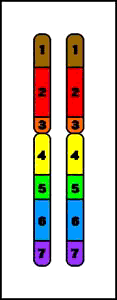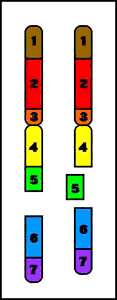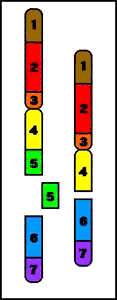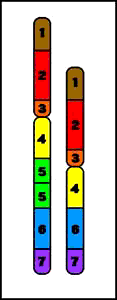
- duplicación de material genético
- conduce a fenotipo anormal
- etiología:
1. de novo - resultado de entrecruzamiento desigual durante la meiosis8. Cromosomas anulares
2. tiene lugar en la descendencia de portadores de translocación- se originan por la pérdida de material genético de amos extremos del cromosoma, con fusión de ambos extremos- inestable durante la mitosis y se puede perder
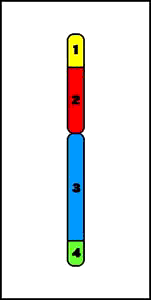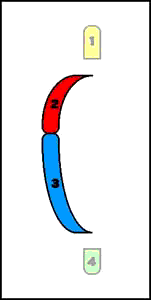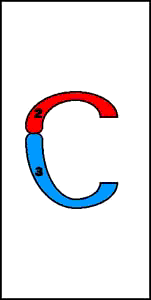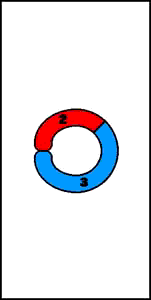
9. Inserciones
1. Individuos con malformaciones congénitas múltiples, retraso mental, fallo de crecimiento2. Individuos con sospecha de anomalías cromosómicas
3. Parejas con pérdidas múltiples durante la gestación
4. Mujeres de corta estatura o amenorrea primaria
5. Varones con infertilidad
6. Pacientes con genitales ambiguos
7. Pacientes con enfermedades hematológicas
1. El cromosoma Filadelfia t(9;22) se encuentra en casi todos los casos de CML (linfoma mieloide crónico ?) 2. La inversión de 16q22 se observa en casi todos los casos de un subtipo de AML (M4Eo)
2. La inversión de 16q22 se observa en casi todos los casos de un subtipo de AML (M4Eo)
| (Fin del documento) | Volver a la 1ª Parte | Volver a la 2ª Parte |