Tema 4
|
|
|
|
) Generalmente
las proteínas se desnaturalizan con calor. Bajo condiciones adecuadas
un incremento mederado de la temperatura conduce a la perdida de la estrutura
secundaria y terciaria de la proteína. Se puede determinar el porcentaje
de moléculas desnaturalizadas a una detarminada temperatura por diferente
técnicas (espectroscopía UV, viscosidad de la disolución
o actividad óptica). Por lo general, la desnaturalización
se produce en un intervalo relativamente estrecho de temperatura. Esto indica
que es un proceso cooperativo donde la perdida de unas pocas intercciones
no covalentes son suficientes para la perdita total de la conformación
nativa. La mayoría de la proteínas poseen una temperatura
de fusión (Tm, temparatura a la cual el 50% de la proteína
se encuentra desnaturalizada) caracteristica a un determinado pH y fuerza
iónica. Generalmente
las proteínas se desnaturalizan con calor. Bajo condiciones adecuadas
un incremento mederado de la temperatura conduce a la perdida de la estrutura
secundaria y terciaria de la proteína. Se puede determinar el porcentaje
de moléculas desnaturalizadas a una detarminada temperatura por diferente
técnicas (espectroscopía UV, viscosidad de la disolución
o actividad óptica). Por lo general, la desnaturalización
se produce en un intervalo relativamente estrecho de temperatura. Esto indica
que es un proceso cooperativo donde la perdida de unas pocas intercciones
no covalentes son suficientes para la perdita total de la conformación
nativa. La mayoría de la proteínas poseen una temperatura
de fusión (Tm, temparatura a la cual el 50% de la proteína
se encuentra desnaturalizada) caracteristica a un determinado pH y fuerza
iónica. |
Desnaturalización de la Ribonucleasa A |
) Los
trabajos de Christian Anfisen con la ribonucleasa A (RNAsa A) apoyan está
idea. La RNAsa A es una enzima digestiva que cataliza la hidrólisis
de ácidos ribonucléicos. Es relativamente pequeña formada
por 124 residuos de aminoácidos y tiene 8 cisteínas que forman
4 puentes disulfuro. Cuando la RNAsa A se desnaturaliza perdiendo su estructura
nativa, y por tanto su actividad, es capaz de recuperar su estructura nativa
y su actividad en determinadas condiciones. La desnaturalización de
la RNAsa A con urea 8 M y B-mercaptoetanol. Reduce los puentes disulfuros
de la enzima. Eliminando la urea y el BME, por ejemplo mediante dialisis,
la enzima recuperasu plegamiento y se forman lo spuentes disulfuros correctos;
la RNAsa A recupera su actividad. Si en cambio solo retiramos el agente reductor.
Los puentes disulfuros que se forman no los los correctos y aproximadamente
solo el 1% de la enzima es activa (si 8 residuos de cisteína forman
puentes disulfuros aleatoriamente, hay 105 posibles combinaciones, 7 para
el primer par, 5 para el segundo, tres para el tercero y solo uno para el
úiltimo, 7x5x3x1 = 105). Si a esta última preparación
se le añaden trazas de BME y se calienta suavemente, los puentes disulfuros
incorrectos se rompen formmandose los adecuados. Los
trabajos de Christian Anfisen con la ribonucleasa A (RNAsa A) apoyan está
idea. La RNAsa A es una enzima digestiva que cataliza la hidrólisis
de ácidos ribonucléicos. Es relativamente pequeña formada
por 124 residuos de aminoácidos y tiene 8 cisteínas que forman
4 puentes disulfuro. Cuando la RNAsa A se desnaturaliza perdiendo su estructura
nativa, y por tanto su actividad, es capaz de recuperar su estructura nativa
y su actividad en determinadas condiciones. La desnaturalización de
la RNAsa A con urea 8 M y B-mercaptoetanol. Reduce los puentes disulfuros
de la enzima. Eliminando la urea y el BME, por ejemplo mediante dialisis,
la enzima recuperasu plegamiento y se forman lo spuentes disulfuros correctos;
la RNAsa A recupera su actividad. Si en cambio solo retiramos el agente reductor.
Los puentes disulfuros que se forman no los los correctos y aproximadamente
solo el 1% de la enzima es activa (si 8 residuos de cisteína forman
puentes disulfuros aleatoriamente, hay 105 posibles combinaciones, 7 para
el primer par, 5 para el segundo, tres para el tercero y solo uno para el
úiltimo, 7x5x3x1 = 105). Si a esta última preparación
se le añaden trazas de BME y se calienta suavemente, los puentes disulfuros
incorrectos se rompen formmandose los adecuados. |
Consideraciones termodinámicas |
) La
conformación nativa de una proteína es la conformación
de más baja energía libre de Gibbs. El proceso de plegamiento,
como cualquier otro proceso biológico, se encuentra bajo control
termodinámico y cinético, y es un proceso que está
claramente favorecido en condiciones fisiológicas. En otras palablas,
el proceso de plegamiento hasta alcanzar la estructura nativa presenta
un ΔG negativo. La
conformación nativa de una proteína es la conformación
de más baja energía libre de Gibbs. El proceso de plegamiento,
como cualquier otro proceso biológico, se encuentra bajo control
termodinámico y cinético, y es un proceso que está
claramente favorecido en condiciones fisiológicas. En otras palablas,
el proceso de plegamiento hasta alcanzar la estructura nativa presenta
un ΔG negativo.Desde el punto de vista entrópico, el proceso de plegamiento supone una disminución neta de entropía desde la estructura denominada ovillo aleatorio a la única estructura nativa. Este descenso de entropía, denominada entropía conformacional, supone un incremento positivo de energía libre en el proceso de plegamiento (ΔG = ΔH – TΔS). Para poder tener un descenso global en el proceso es necesario que el incremento de entalpía sea negativo o que existan otros aumentos de entropía. La principal contribución entalpica al proceso de plegamiento la constituyen la formación de interacciones no covalentes que estabilizan la estructura nativa y las interacciones hidrofóbicas entre las cadenas laterales apolares que generalmente quedan localizados en el interior de la estructura nativa. |
En resumen, la estabilidad de la estructura plegada de una proteína
globular depende de la interacción de tres factores:
|
| Parametros termodinamicos de algunas proteínas globulares a 25°C en disoluciones acuosas | |||
| Proteína | ΔG (kJ/mol) |
ΔH (kJ/mol) |
ΔS (J/K·mol) |
| Ribonucleasa | -46 |
-280 |
-790 |
| Quimiotripsina | -55 |
-270 |
-720 |
| Lisozima | -62 |
-220 |
-530 |
| Citocromo c | -44 |
-52 |
-27 |
| Mioglobina | -50 |
0 |
+170 |
| Cada dato se ha tomado al pH en el que la proteína presenta máxima estabilidad (todos cerca del pH fisiológico). Los datos son para el equilibrio de plegado: desnaturalizada ->nativa. | |||
La paradoja de Levinthal |
| El plegado de proteínas globulares a partir de sus conformaciones desnaturalizadas
es un proceso notablemente rápido, que se completa en menos de un segundo.
Esto puede resultar paradójico tal y como puso de manifiesto Cyrus Levinthal
en 1968. Una proteína pequeña como la RNAsa A, que tiene 124 aminoácidos
tiene 1050 conformaciones posibles. Si la molécula pudiese
probar una configuración cada 10-13 segundos, serían
necesarios 1030 años para probarlas todas. Sin embargo, se
ha comprobado in vitro que la RNAsa de pliega en aproximadamente 1
minuto. La salida al dilema de Levilthal podría estar en la selección acumulativa, según la cual los intermediarios parcialmente correctos se retienen. Los sitios de iniciación favorecen la estabilización de estructuras más complejas. |
Determinados residuos aparecen preferentemente en determinadas estructura secundarias |
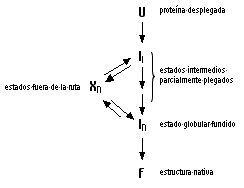 Actualmente no se conoce el proceso exacto mediante el cual se produce
el plegamiento “de novo” de una proteína. Se admite que
este proceso se inicia con interaciones de corto alcance que forman estructuras
secundarias en regiones locales del polipéptido. Estas interacciones
son interacciones no covalentes que se establecen entre las cadenas laterales
próximas. Determinados residuos tienen tendencia a formar estructuras
en hélice-alfa, hoja-beta o giros reversos (ver tabla). Estos residuos
que se estructuran espontaneamente se conocen como sitios de iniciación. Actualmente no se conoce el proceso exacto mediante el cual se produce
el plegamiento “de novo” de una proteína. Se admite que
este proceso se inicia con interaciones de corto alcance que forman estructuras
secundarias en regiones locales del polipéptido. Estas interacciones
son interacciones no covalentes que se establecen entre las cadenas laterales
próximas. Determinados residuos tienen tendencia a formar estructuras
en hélice-alfa, hoja-beta o giros reversos (ver tabla). Estos residuos
que se estructuran espontaneamente se conocen como sitios de iniciación.El siguiente paso es la formación de estructuras parcialmente plegadas que se conocen con el nombre de estado globular fundido. Este estado se produce tras un colapso hidrofóbico, y contiene la mayor parte de las estructuras secundarias presentes en la estructura nativa, pero posee muchas interacciones incorrectas. Este estado se encuentra en rápido equilibrio con el estado de ovillo aleatorio totalmente desnaturalizado. Las interacciones de medio y largo alcance se forman mediante reordenaciones del estado globular fundido. Las últimas interacciones en formarse son los puentes disulfuros. |
| Frecuencias relativas
de los residuos de aminoácido en estructuras secundarias
|
||||
| Aminoácido | Hélice
alfa |
Hoja
beta |
Giro |
Preferencia
por |
| Alanina | 1,29 |
0,90 |
0,78 |
hélices
alfa
|
| Cisteína | 1,11 |
0,74 |
0,80 |
|
| Leucina | 1,30 |
1,02 |
0,59 |
|
| Metionina | 1,47 |
0,97 |
0,39 |
|
| Glutamato | 1,44 |
0,75 |
1,00 |
|
| Glutamina | 1,27 |
0,80 |
0,97 |
|
| Histidina | 1,22 |
1,08 |
0,69 |
|
| Lisina | 1,23 |
0,77 |
0,96 |
|
| Valalina | 0,91 |
1,49 |
0,47 |
hojas betas
|
| Isoleucina | 0,97 |
1,45 |
0,51 |
|
| Fenilalanina | 1,07 |
1,32 |
0,58 |
|
| Tirosina | 0,72 |
1,25 |
1,05 |
|
| Tripofano | 0,99 |
1,14 |
0,75 |
|
| Treonina | 0,82 |
1,21 |
1,03 |
|
| Glicina | 0,56 |
0,92 |
1,64 |
giros
|
| Serina | 0,82 |
0,95 |
1,33 |
|
| Aspartato | 1,04 |
0,72 |
1,41 |
|
| Asparragina | 0,90 |
0,76 |
1,28 |
|
| Prolina | 0,52 |
0,64 |
1,91 |
|
| Arginina | 0,96 |
0,99 |
0,88 |
|
Predicción de la estructura de las proteínas |
| Dado que determinados residuos presentan una mayor frecuencia relativa
de aparición en las estruturas secundarias y que la información
necesaria para el correcto plegado de una proteína parece estar contenida
en su estructura primaria, uno de los grandes retos de la Bioinformática
es la predicción de la estructura terciaria de las proteínas.
La predicción de la estructura a partir de la secuencia de amnoácidos ha resultado difícil, fundamentalmente debido a las interacciones de largo alcance que estabilizan las estructuras secundarias y terciaria. Para intentar de resolver este problema se han tomado dos aproximaciones experimentales diferentes: la predicción ab initio, que intenta resolver el problema sin tener en cuenta ninguna de las estructura conocidas; y el modelado por comparación (diseño de proteínas por homología), que realiza la predicción en función de un modelo conocido. Este último método asume que la estructura está más conservada que la secuencia, de modo que si la proteína que se quiere modelar presenta más de un 30% de identidad con una proteína de estructura conocida, ambas proteínas serán estructuralmente semejantes. La mayoría de los métodos para la predicción de la estructura secundaria de las proteínas comienza con el alineamiento de la secuencia que se pretende modelizar. Disponer de la estructura tridimensional de una proteína para el análisis de su función es indudablemente un valor añadido. A pesar del avance en las técnicas experimentales para proporcionar modelos aproximados de la estructura y la dinámica de las proteínas (cristalografía de rayos X o resonancia magnética nuclear), cada día aumenta la diferencia entre el número de secuencias y el de estrucuras conocidas. Actualmente se emplea la Bioinformática para la predicción de la estructura de las proteínas. Los métodos de predicción de estructuras tiene por objetivo disminuir esta diferencia entre el número de secuencias conocidas y el de estructuras. |
Plegamiento asistido de proteínas |
| A pesar de que la estructura primaria de la proteínas contiene
la información necesaria para el correcto plegamiento de la misma,
muchas proteínas necesitan el concurso de otras proteínas
para logar su conformación nativa, y por tanto su conformación
funcionalmente activa. estas proteínas que facilitan el correcto
plegamiento de otras son la proteína disulfuro isomerasa, peptido
prolil cis-trans isomerasa y las chaperonas moleculares. La proteína disulfuro isomerasa cataliza el intercambio o la mezcla de puentes disulfuros hasta que se forman los enlaces de la estructura nativa. la péptido prolil cis-trans isomerasa cataliza la interconversión de los isómeros cis-trans en los enlaces peptídicos de prolina. |
|
|
|
|
Última actualización:
28-Jun-2005
Comentarios y sugerencias: José
Luis Urdiales
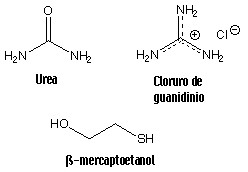 La estructura
tridimensional de una proteína en condiciones fisiológicas
se conoce como estructura nativa, y se considera la estructura
más estable de todas las estructuras posibles. Además la estructura
nativa es la funcionalmente activa. Si cambiamos las condiciones ambientales,
la estructura nativa se pierde, este proceso se denomina desnaturalización.
Podemos conseguir la desnaturalización de una proteína mediante
anumento de la temperatura, cambios drásticos de pH, añadiendo
agentes caotrópicos como la urea o el clorhidrato de guanidinio)
o mediante disolventes orgánicos como el etanol.
La estructura
tridimensional de una proteína en condiciones fisiológicas
se conoce como estructura nativa, y se considera la estructura
más estable de todas las estructuras posibles. Además la estructura
nativa es la funcionalmente activa. Si cambiamos las condiciones ambientales,
la estructura nativa se pierde, este proceso se denomina desnaturalización.
Podemos conseguir la desnaturalización de una proteína mediante
anumento de la temperatura, cambios drásticos de pH, añadiendo
agentes caotrópicos como la urea o el clorhidrato de guanidinio)
o mediante disolventes orgánicos como el etanol.